
易基因微生物组学测序分析成果见刊《Front Microbiol》
2023年02月17日,中国农业科学院深圳农业基因组研究所Adnan Khan、云南省第一人民医院米弘瑛为共同第一作者,中山大学附属第六医院郝虎/李思涛、南方医科大学附属佛山市妇幼保健院戴怡蘅为论文共同通讯作者在《Front Microbiol》杂志以“Dynamic changes of the gut microbial colonization in preterm infants with different time points after birth”为题发表研究论文。该研究通过微生物组学的16s RNA等实验揭示了12名早产儿在出生后第1、7、14、21、28、42天六个时间点(C1、C2、C3、C4、C5、C6)肠道微生物定植过程中的动态变化,为早产儿在出生后不同时间点针对性细菌治疗提供新的视角。深圳易基因科技为本研究提供微生物组测序分析服务。

标题:Dynamic changes of the gut microbial colonization in preterm infants with different time points after birth 早产儿出生后不同时间点肠道微生物定植的动态变化
时间:2023-02-17
期刊:Frontiers in Microbiology
影响因子:IF 6.064
技术平台:16S rRNA等

摘要:
与早产相关的风险在所有妊娠中分布不均衡。在孕早期,坏死性小肠结肠炎(NEC)和晚发性败血症(LOS)等并发症更为常见,且与肠道微生物组组成变化相关。常规细菌培养技术表明,早产儿肠道微生物组的定殖与健康足月儿的肠道微生物组定殖有显著差异。本研究旨在分析早产儿在不同时间点(出生后第1、7、14、21、28、42 天)粪便微生物组的动态变化。研究招募了2017年1月至2017年12月在中山大学附属第六医院住院的12名早产儿,利用16S rRNA基因测序分析对130份早产儿粪便样本进行分析。研究结果表明早产儿粪便微生物群的定植过程在出生后的不同时间点呈高度动态变化,即Exiguobacterium、Acinetobacter和Citrobacter随着年龄增长表现出丰度下降模式,而在42天的早产儿粪便微生物群发育过程中,肠球菌(克雷伯氏菌和大肠杆菌)菌群逐渐生长成为主要微生物群。而早产儿肠道双歧杆菌的定植相对较晚,并没有迅速成为主要的微生物群。此外,研究结果还表明Chryseobacterium菌群的存在,且在不同时间点的定殖不同。本研究为早产儿出生后不同时间点针对性细菌治疗提供新的视角。
材料和方法:
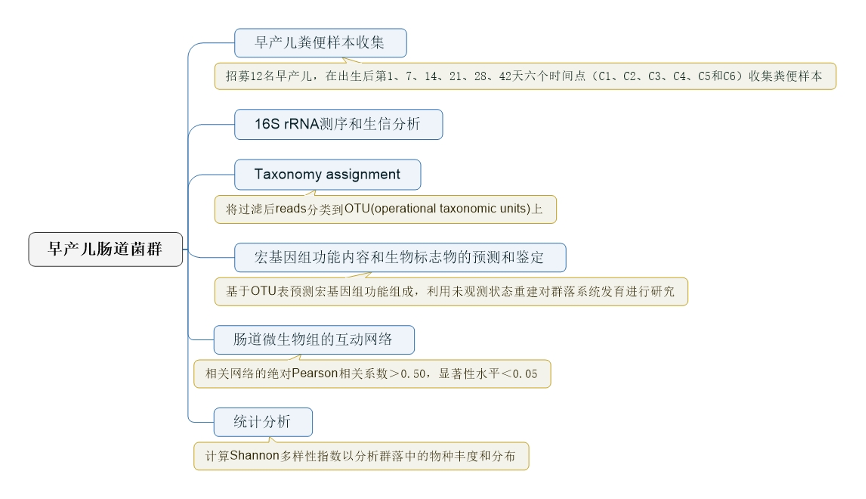
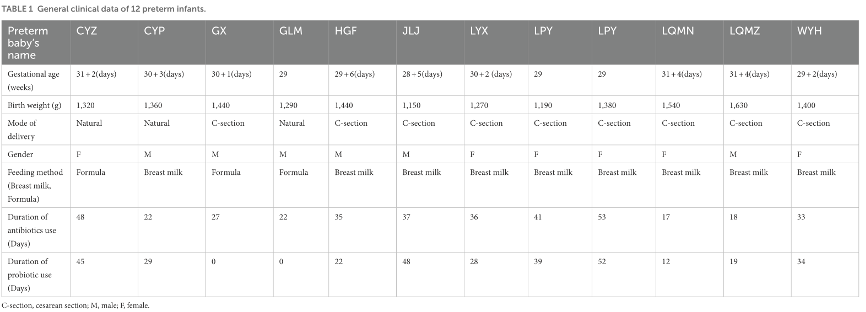
表1:12例早产儿的一般临床数据
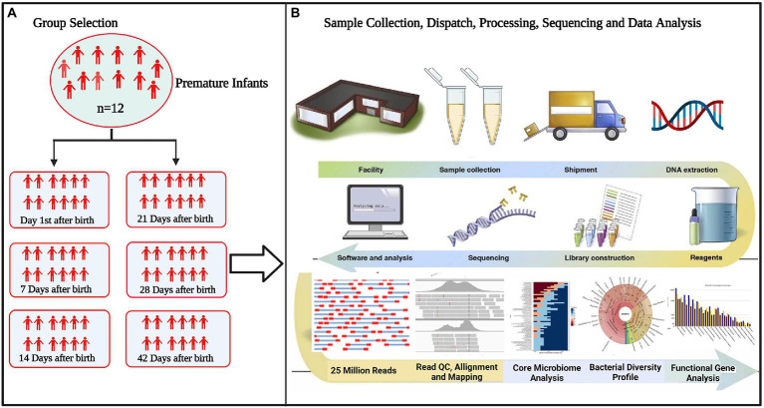
图1:研究流程图
A. 选择实验组以鉴定组间微生物群的差异
B. 标准化技术因素和样品处理,以控制过程的每个步骤引入的变化。从临床变量到样本处理,收集和管理每个样本各个方面的metadata做数据分析。
实验结果
(1)早产儿微生物群落组成的综合表征
研究人员使用来自12名早产儿出生后多个时间点(C1-C6)的130份粪便样品的16S rRNA基因数据来评估微生物多样性的时间相关变化,鉴定不同时间点微生物组中的最大丰度和富集驱动因子,并分析粪便微生物在早产儿健康发育过程中的表型和功能。
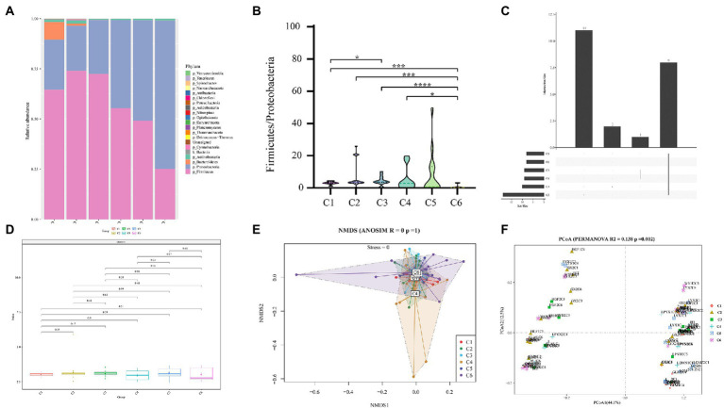
图2:早产儿6个时间点肠道微生物类群相对丰度的比较。
A. 从门(Phyla)水平上分析6个时间点肠道微生物群的组成。
B. 组间(C1–C6)的F/P比值箱形图。
C. 六个时间点组间肠道微生物门(Phyla)的重叠分析Venn图。包括在六个以上的粪便样本中检测到的门。
D. 使用Shannon指数分析六组细菌群落的α多样性和丰度。
E. 使用Bray–Curtis差异的NMDS进行Beta(β)多样性分析。
F. 不同时间点组间肠道微生物群的未加权Unifrac距离(Unweighted Unifrac distance)。使用非参数Kruskal–Wallis检验和Tukey检验计算成对p值:*p<0.05、**p<0.01、***p<0.001、****p<0.0001为具有显著性。
(2)早产儿出生后不同时间点肠道菌群中丰度最高的属(genera)
对丰度最高的属分析与β多样性分析结果一致,表明丰度最高的属多样性随时间推移而增加。结果表明丰度最高的属(Exiguobacterium、Prevotella、Acinetobacter、Pseudomonas、Enterococcus、Bifidobacterium、Escherichia-Shigella、Klebsiella、Gardnerella、Streptococcus和Chryseobacterium)发生了时间依赖性变化,且在各组区系结构中的样本异质性增加。图3A显示了不同时间点组中前40个丰度最高的属,其中一半为所有时间点组间共有(图3B)。图3C中的热图通过聚类分析表明,C1-C3中的粪便微生物群由Exiguobacterium、Pseudomonas、Lactococcus、Brevundimonas、Burkholderiaceae和Thermus组成;C3-C5中为Enterococcus富集区,在C6中为Escherichia−Shigella富集区。
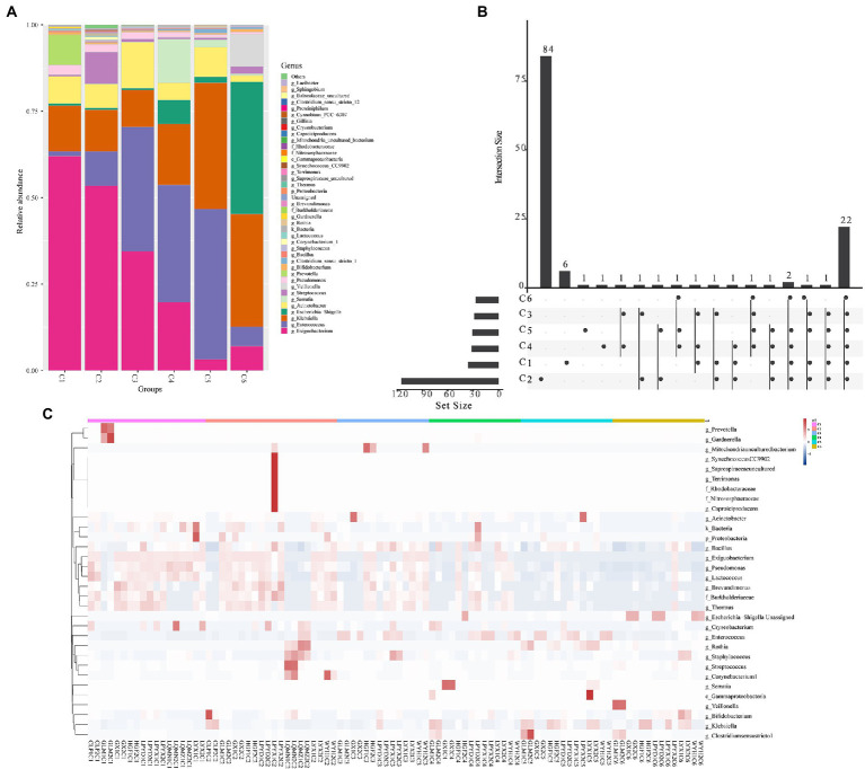
图3:丰度最高的肠道微生物属在早产儿6个不同时间点组中富集情况。
A. 6个时间点(C1-C6)内丰度最高的前40个属
B. 6个不同时间点组间肠道微生物属的重叠分析Venn图。
C. 6个时间点组间肠道微生物群中差异表达最显著的属。名称前面的单个字母(g、f和c)分别表示属、门和纲。
(3)早产儿6个时间点肠道微生物组的差异分类
为进一步表征六个时间点组间肠道微生物群的动态变化,本研究在不同的分类水平上对这些分类群的系统发育变化进行时间点依赖性分布分析,即:属、门、纲、目、科、种。
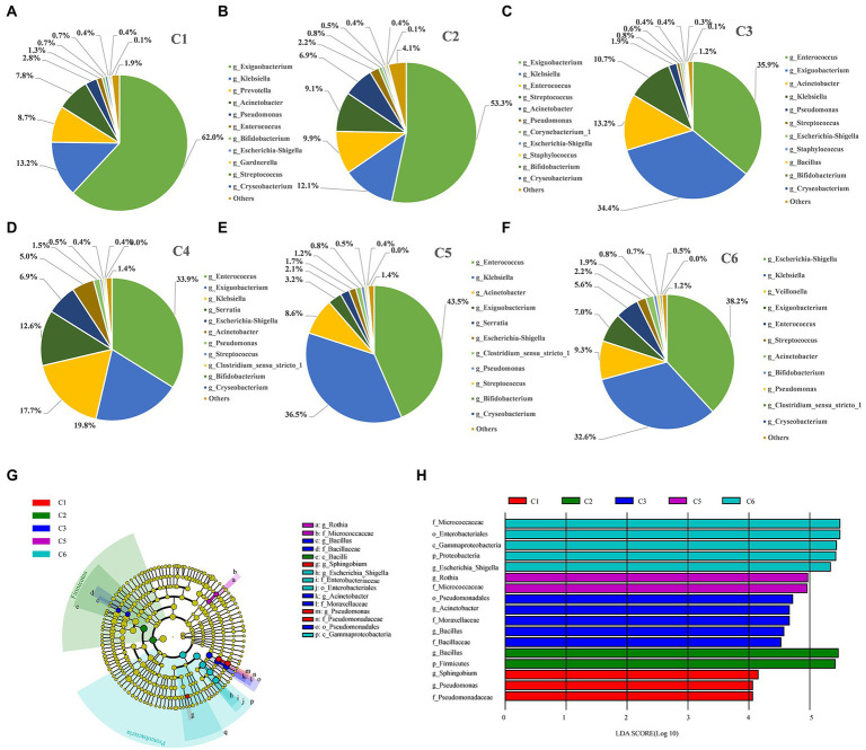
图4:在早产儿的六个时间点组中富集的显著差异分类群。
A-F. 早产儿不同时间点肠道优势微生物属(genus)水平的动态变化。
A. 从门到属水平的显著差异分类群系统发育分支图。微生物类用字母表示。每个节点表示不同分类级别的一个分类单元。节点颜色是在相关队列(C1-C6)中观察到的丰度较高的分类群。
B. 具有显著差异的物种LDA得分大于预估值;默认分数为2.5。直方图长度表示LDA分数;表示不同群体之间具有存在显著差异的物种影响影响程度。
(4)早产儿年龄依赖性肠道微生物群网络及其关键驱动属
研究人员利用稀疏成分相关性(SparCC)分析来研究所有时间依赖性样品中肠道微生物之间的相互作用。所有相对丰度≥0.1%的属都包含在网络中。Exiguobacterium(Firmicutes)网络与不动杆菌(Proteobacteria)、假单胞菌(Proteobacteria)和乳球菌(Firmicutes)的相关性最强。同样,Serratia(Proteobacteria)也与Veillonella(Firmicutes)呈现出强相关性。但Streptococcus、Staphylococcus、Enterococcus、Bacillus (Firmicutes)与其他属的相关性最弱。前12个显著差异属的热图和层次聚类分析表明了样本的相关性(|FC|>1、p<0.05)。
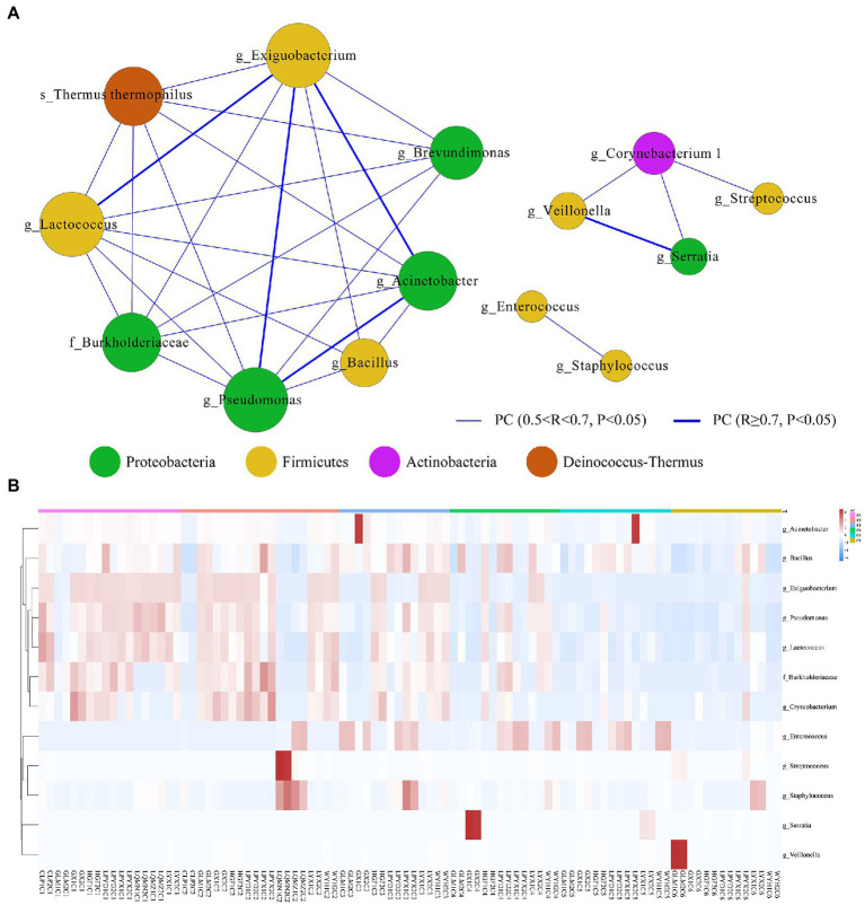
图5:早产儿年龄依赖性肠道微生物群网络及其关键驱动属
A. 根据SparCC结果构建早产儿的微生物交互网络,并使用Cytoscape进行可视化。(C1)出生后第1天、(C2)出生后第7天、(C3)出生后第14 天、(C4)出生后第21天、(C5)出生后第28天、(C6)出生后第42天。网络包含平均丰度>0.1%的属、相关性|R|>0.2和p<0.05,节点颜色表示属的门,节点大小表示加权节点连接,边缘颜色和厚度表示相关性。
B. 所有时间依赖性样本(C1–C6)中前12个显著差异的微生物属热图。红色对应于上调的基因产物,绿色对应于下调的基因产物。行表示每个差异表达的门,列表示每个样本
(5)基于分类组成预测微生物代谢功能
为更好了解早产儿肠道微生物组、疾病易感性和细菌代谢的功能差异,研究人员利用PICRUSt通过分析生成的OUT及其参考基因组数据库的16S rRNA数据中的基因,来预测细菌宏基因组的基因家族。结果表明,在早产儿C1、C2、C3、C5和C6组中,分别有63个、1个、5个、22个和24个KEGG通路显著过表达。C1、C2、C3和C5组中鉴定的大多数通路为维持生命所必需,包括ABC转运蛋白、核苷酸代谢、抗坏血酸、醛酸盐代谢、碳水化合物、蛋白质代谢二恶英降解和脂质代谢。描述代谢过程和免疫状态的许多通路在所有组中均过度表达(over-represented,即富集enriched)。所有这些通路对于影响微生物在环境中的分布、存活和增殖至关重要,结果还需要使用宏基因组学来进一步证实。
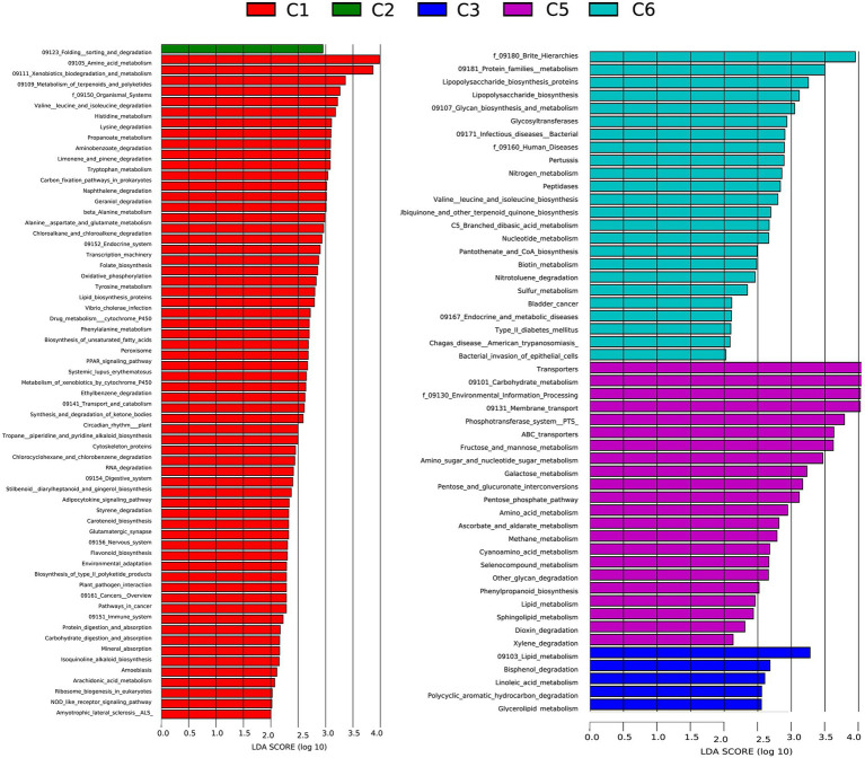
图6:早产儿出生后6个时间点(C1、C2、C3、C5、C6)的组间功能模块相对丰度比较,使用从PICRUSt生成的线性判别分析效应量(LEfSe)分析。35个、1个、5个、18个和15个KEGG通路分别在C1、C2、C3、C4和C6中显著富集。数据显示,所有组间预测的细菌代谢功能存在差异。LDA评分>2.5表示具有显著差异的通路。
结论:
本研究结果将有助于更好地了解早产儿出生后肠道微生物群变化对健康的长期影响,本研究首次对出生42日龄以内的早产儿粪便微生物组组成进行研究。结果表明早产儿粪便微生物群在出生后一天左右较为简单,Exiguobacterium、Acinetobacter和Citrobacter占整个微生物群的83%。随着时间的推移,这三个主要细菌家族的相对丰度下降,而Enterococcus、Klebsiella、Escherichia菌群逐渐增加,并成为主要菌群,其中任何一种细菌都可能引起消化道感染。同时,早产儿肠道双歧杆菌的定植相对较晚,并没有迅速成为主要的微生物群。而新生儿重症监护室早产儿的粪便微生物群中存在Chrysobacterium菌群,表明其健康存在额外风险。本研究旨在为比较和理解早产儿肠道微生物群的时间依赖性动态变化开辟新的可能性,
并为早产儿在出生后不同时间点针对性细菌治疗提供新的视角。
参考文献:https://www.pmop.cn/pubmed/36876108
相关阅读:
IF14 项目文章 | 东北农业大学张志刚团队:基于多组学分析噻虫啉暴露对日本鹌鹑微生物-肠-肝轴的影响
项目文章 | 90天见刊,易基因m6A RNA甲基化(MeRIP)+转录组组学研究
Nature | DNA甲基化在胚胎全能8细胞人工诱导过程中的时空演绎
项目文章 | 组蛋白ChIP-seq揭示烟粉虱共生菌Hamiltonella调控宿主生殖新机制


